Summary:
Fungal infections have constantly increased over the last few decades, resulting in 1.5 million deaths worldwide per annum. Currently available antifungal drugs are no longer reliable due to poor efficacy, host toxicity and other regulatory restrictions. A class of cyclic lipopeptides called octapeptins have shown promising results against several microbial species. However, the complete antimicrobial spectrum of these compounds are yet to be discovered. In this regard, octapeptin C4, a subclass of the octapeptin family was synthesized along with its two analogues with new lipophilic groups: one from the echinocandin class of antifungal drug Micafungin, the other from a novel lipopeptide antibiotic, MX-2401. The yields of pure natural octapeptin C4 and Micafungin and MX-2401 analogues were 18.0%, 26.0% and 15.0% respectively. Followed by synthesis, in vitro assay of these compounds was performed against Cryptococcus neoformans.
1. Introduction:
1.1 Clinical Background:
Fungal infections have drastically increased since 1960s, affecting 1.2 billion people [1, 2]. These infections are prevalent worldwide, threatening lives of various immunocompromised patients (cancer, HIV/AIDS etc.) [3, 4]. Around 1.5 million people per annum die of invasive fungal infections; 90% of these are caused by the following fungal species: Aspergillus, Candida, Pneumocystis, Cryptococcus, Rhizopus and Mucor [4].
Fungal pathogens are often difficult to treat in mammalian hosts due to their similar eukaryotic cellular composition. Due to unavailability of fungal specific drug targets, there is always a high chance of host toxicity. Therefore, discovery of new, selective antifungal therapies is challenging [1, 4-6]. Options to treat fungal infections have been limited to the following: (1) membrane function disruptors (amphotericin B); (2) agents inhibiting DNA/RNA synthesis (flucytosine); (3) ergosterol synthesis inhibitors (azoles) and (4) glucan synthesis inhibitors (echinocandins) [7]. However, these current therapies are hindered by poor efficacy, side effects and toxicity [4, 8]. Therefore, it is necessary for scientists to search for alternative therapies to combat fungal infections.
Cryptococcosis is a type of systemic mycosis, caused by Cryptococcus neoformans [9]. C. neoformans is a pathogenic fungus responsible for deadly mycotic infections in AIDS patients, particularly in sub-Saharan Africa [10, 11]. These infections can further lead to encephalitis, meningitis or meningoencephalitis, which are difficult to treat and need proper medical attention [9, 12].
The polysaccharide capsule of C. neoformans is negatively charged [13], and has a potential role in maintaining its virulence [14, 15]. It guards the fungal cell against phagocytosis and facilitates opportunistic infections [15, 16]. In addition, the capsule enables the fungal cell to disseminate by both surviving and replicating in the lysosome [17]. Therefore, due to the capsule and a variety of other survival mechanisms [18, 19], cryptococcal infections can rapidly spread through the body and eventually reach the brain, causing cryptococcal meningitis [20].
According to WHO guidelines, the optimal treatment for cryptococcal meningitis begins with a two week course of amphotericin B along with flucytosine, followed by consolidation therapy with fluconazole [21]. However, flucytosine is expensive and unlicensed in many regions, like sub-Saharan Africa [21, 22]. In this case, therapy is confined to amphotericin B and fluconazole, or fluconazole monotherapy if amphotericin B is unavailable [22]. Unfortunately, a number of fluconazole resistant cases have been reported in recent years, which has made the fight against cryptococcal meningitis more urgent [22].
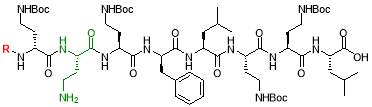
In 1970s, a class of cyclic lipopeptides called ‘octapeptins’ were discovered which showed efficacy against various microbial species, but little research has been made to explore its full antimicrobial spectrum [13]. Octapeptin C4 is a member of the octapeptin class, having a 3-hydroxy-decanoic acid moiety attached to the core structure [13, 23] (Fig. 2). It has been proposed that the negatively charged capsule of C. neoformans can concentrate cationic lipopeptides in the cell surface, thereby increasing fungal sensitivity to octapeptin C4 [13]. In addition, SAR of nine alanine scan derivatives of octapeptin C4 have revealed that the lipophilic groups as well as the cationic amino acid moieties are important for its antifungal properties [13]. To study the interaction between fungal capsule and octapeptin C4, it is desirable to synthesize analogues of these compounds, followed by in vitro assay against C. neoformans. The synthesis of these compounds can be achieved using a suitable technique like Solid Phase Peptide Synthesis (SPPS).
1.2 An overview of the Solid Phase Peptide Synthesis:
The importance of synthetic peptides in research and as therapeutics can hardly be denied. Synthesis of biologically active peptides provides opportunities to obtain novel compounds with improved biological properties and paves the way to study structure-activity relationships. Recombinant technologies are used to synthesize large peptides, whereas Solid Phase Peptide Synthesis (SPPS) is the most feasible approach for the synthesis of small peptides [24].
The major theme for SPPS is based on the elongation of a peptide chain in multiple steps, where the first amino acid is covalently linked to a solid support (resin) via a linker [25, 26] (Fig. 1). After the first amino acid is coupled to the resin, the α-amino protecting group is removed, followed by the addition of the second amino acid, resulting in the formation of a peptide bond [24, 25]. In this process, a series of amino acids are added to the growing peptide chain in a stepwise progression, involving a number of repeated deprotection and coupling reactions until the target peptide is assembled [25]. Finally, the peptide is cleaved off the resin, isolated and purified using suitable techniques [24, 25].
SPPS has got some major advantages over other conventional synthetic techniques [24, 25] as follows:
- A simplified and accelerated multistep synthesis, providing the reaction to be completed in a single reaction vessel
- The use of excess amino acids drives the reaction to full completion with minimal chance of product loss, thereby minimizing the need for purification of each intermediate product
- High yield of the final product since excess reagents are used to ensure full completion of each reaction step





Solid support (Resin)




Amino acids


📝
Need a custom essay written to your exact brief?
Our PhD-qualified writers craft original, plagiarism-free essays tailored to your topic, word count, citation style, and deadline — argumentative, analytical, descriptive, or reflective. Every paper is 100% written from scratch.
✓ Plagiarism-free · ✓ 100% human-written · ✓ Free revisions · ✓ Confidential
🔒 No payment to start · From $11/page

Protecting groups


















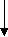
Cleavage from resin, isolation, purification
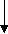





Fig. 1. Diagrammatic representation of SPPS (Adapted from Merrifield, R. B. [27])
1.3 Fmoc SPPS vs. Boc SPPS:
In order to protect the amino acids, two strategies are widely used: (a) the butyloxycarbonyl (Boc)-protecting group or (b) 9-fluorenyl methoxy carbonyl (Fmoc) group [24]. The Boc protecting group is acid-labile and protects the α-amino group whereas the side chains are protected by orthogonal protecting groups such as benzylic groups. The Boc and benzylic groups can be cleaved by trifluoro acetic acid (TFA) and hydrogen fluoride (HF) respectively. On contrary, the Fmoc protecting group is base-labile, protecting the α-amino group, but the side chains are protected by acid labile tert-butyl or trityl-based groups. Fmoc can be cleaved by 20% piperidine in dimethylformamide (DMF), whereas removal of tert-butyl groups is achieved by TFA [24].
✍️
Your thesis deserves more than a rushed draft.
Master's and doctoral thesis writing requires a defensible argument, rigorous methodology, and impeccable academic writing. Our thesis specialists have delivered 8,400+ model documents that passed committee review.
✓ Plagiarism-free · ✓ 100% human-written · ✓ Free revisions · ✓ Confidential
🔒 No payment to start · From $11/page
Fmoc-SPPS is preferred to Boc-SPPS, since the later involves use of HF which is hazardous, and the TFA used for Boc-removal may result in peptide loss at each synthetic step. Fmoc deprotection releases a fluorene group which has strong UV absorption properties. Therefore, Fmoc-SPPS is considered as a truly orthogonal protecting strategy for the production of smaller peptides [24, 28].
1.4 Objectives of the research project:
Previously, SPPS was applied to synthesize natural octapeptin C4 and other alanine scan derivatives [13]. Therefore, synthesis of octapeptin C4 and analogues will provide an opportunity to further characterize these compounds by structure activity relationship (SAR) studies.
This research project aims to investigate the following:
- Synthesis of octapeptin C4 and analogues with novel lipid groups using “Fmoc Solid Phase Peptide Synthesis” (Fmoc-SPPS). Two novel lipid groups were used to synthesize octapeptin C4 analogues: one from the echinocandin class of antifungal drug Micafungin, the other from a novel lipopeptide antibiotic, MX-2401 (Fig. 2).
- In vitro assay of octapeptin C4 and analogues against C. neoformans.
Fig. 2. Different octapeptin C4 analogues
2. Materials and Methods:
2.1 Synthetic route to octapeptin C4:
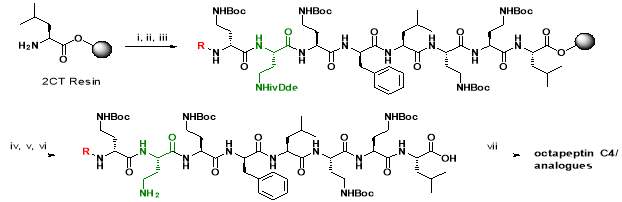
Scheme 1: Solid phase synthesis and off-resin cyclisation of octapeptin C4. Reagents: i) amino acids, HBTU, DIPEA, DMF; ii) 20% piperidine in DMF; iii) Lipophilic moiety (R1,2,3), HBTU, DIPEA, DMF; iv) 4% hydrazine hydrate in DMF; v) 20% HFIP in DCM; vi) DPPA, DIPEA, DMF; vii) TFA/H2O/TIS
Octapeptin C4 and analogues were synthesized by Fmoc-SPPS, in a route developed by Becker et al. [23]. The peptide was synthesized on a 2-chlorotrityl (2-CT) resin, using Fmoc-protected amino acids on the N-terminal. The amide side chains on 2, 4-diaminobutyric acid (Dab) were protected by a Boc group. Orthogonal ivDde protection on Dab (marked green in Scheme 1) allowed the amino group to be selectively deprotected without affecting other Boc-protected groups [23]. A unique feature of ivDde is that, it is stable to 20% piperidine in DMF but cleaved by 4% hydrazine hydrate in DMF [29]. This enables the amino groups of ivDde-protected residues to be selectively deprotected without affecting side chain protecting groups on the other residues. In addition, the deprotection reaction creates a UV-active indazole by-product, which can be easily monitored by spectrophotometry [29].
📚
Tight deadline? We've handled tighter.
Our fastest writers turn around quality academic work in 3 hours. Full essay, research paper, case study — delivered before your submission time.
24/7 support · No hidden fees
Followed by orthogonal ivDde protection, the resin was divided and lipid moieties (R1,2,3, Fig. 2) were added. The ivDde protecting group was cleaved by 4% hydrazine, followed by resin cleavage with a solution of 20% hexafluoroisopropanol (HFIP) in dichloromethane (DCM). Cyclization of the peptide was achieved using diphenyl phosphoryl azide (DPPA), diisopropylethylamine (DIPEA) base in dimethylformamide (DMF). Global deprotection using trifluoroacetic acid (TFA) and purification by HPLC yielded the final octapeptin C4 analogues [23].
2.1.1 Fmoc Solid phase peptide synthesis (Fmoc SPPS):
i) Amino acids, HBTU, DIPEA, DMF

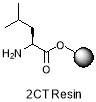
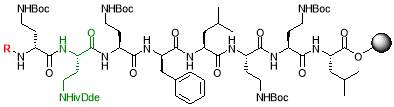
ii) 20% piperidine in DMF
iii) Lipophilic moiety (R1,2,3),HBTU, DIPEA, DMF
The 2-CT resin with L-leu (1.5 gm, 1.08 mmol, loading = 0.72 mmol/g) was suspended in DMF (4 mL). The Fmoc-protected amino acid (1 equiv.) was dissolved in DMF (2 mL) and treated with DIPEA (356 µL, 4 equiv.) and a solution of HBTU in 0.5M DMF (2.16 mL, 2 equiv.). The solution was agitated for 1 min and added to the suspended resin. The reaction was shaken for 2 hours, drained, washed with DMF (3 x 2 mL), methanol (3 x 2 mL) and DCM (3 x 2 mL) and left to dry in vacuo for 15 mins. The entire procedure was repeated with an additional equivalent of amino acid to ensure complete coupling. Finally, the resin was treated with 20% piperidine in DMF (4 mL) and agitated for 30 mins to remove the Fmoc protecting group, so that the next amino acid can be added to the growing peptide chain. The resin was drained, and washed as before with DMF, methanol and DCM [23]. This procedure was repeated to couple each amino acid. After completion of peptide synthesis, the resin was divided and all lipid groups (R1,2,3) were added (1.5 equiv.) using the similar protocol as mentioned.
In order to check the coupling efficiency at each synthetic step, 10 µl of 95: 5: 5 TFA/H2O/TIS was added to a small amount of resin (~5 mg) and kept for 10 mins to clear the peptide from the resin, followed by addition of 50: 50 acetonitrile/H2O (1 mL), filtered and finally LC-MS was performed [23].
2.1.2 Deprotection of ivDde from resin:
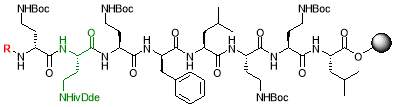
4% hydrazine hydrate in DMF

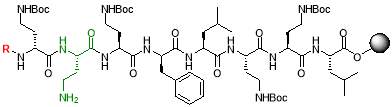
The resin (250 mg, 0.18 mmol) was swollen in THF (5 mL) for 30 mins and drained under vacuum. A solution of 4% hydrazine hydrate in anhydrous DMF (3.25 mL, 8 equiv.) was added to the resin and agitated for 1 hour. The solvent was drained, and washed with DMF (3 x 2 mL), methanol (3 x 2 mL) and DCM (3 x 2 mL). The resulting resin was dried under vacuum overnight [23].
2.1.3 Peptide cleavage from resin:
20% HFIP in DCM
The resin (250 mg, 0.18 mmol) was treated with a cleavage solution, comprising of 20% HFIP in DCM (1:4, 20 mL) for 1 hour. The resulting suspension was drained and resin was shaken with an additional volume of cleavage solution (10 mL) for 30 mins. The solvent was drained and washed with DCM (3 x 3 mL). The filtrates were combined and concentrated in vacuo to afford the crude product [23].
2.1.4 Cyclization of the peptide:
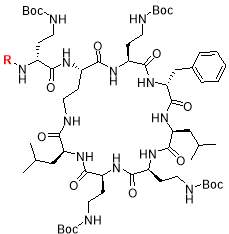
DPPA, DIPEA, DMF
The cleaved peptide was treated with DMF (8 mL), DPPA (4 equiv.) and DIPEA (8 equiv.) and stirred at 60ºC overnight for cyclization. All compounds were purified directly from reaction mixture using GRACE® Reveleris medium pressure liquid chromatography (MPLC) system with C-18 cartridge and acetonitrile/H2O as mobile phase [23]. After LC-MS, all appropriate fractions were combined and freeze dried to give final products (Table 1).
Table 1. LC-MS analysis of the cyclized peptide
| Compound with lipid group | Amount (mg) | Retention time* |